Reducing Risks and Delays in the Translation of Cell and Gene Therapy Innovations into Regulated Products

Introduction
Cell-, gene-, and tissue-based advanced therapeutic products, referred to in this paper as “cell and gene therapy” (CGT) products, have the potential to address many unmet medical needs. These products are commonly discovered and developed in academic laboratories or small biotechnology companies with expertise in science and innovation but limited experience in bringing these products to market. As a result, the process may include avoidable risks that lead to unnecessary costs and delays later in development.
This paper seeks to inform researchers and early developers of potential risks, provide insights on how to minimize these risks, and outline a pathway for easier translation of research into later-stage product development and commercialization. This paper will not be comprehensive. However, it will discuss common but avoidable problems related to pharmaceutical quality and manufacturing control, nonclinical safety testing, intellectual property, communication, and technology transfer. Whereas a product may come to market in various regions of the world, global regulatory requirements should be considered when developing CGT products. Table 1 provides an overview of the key points from this paper. A list of relevant legislation and guidance from the European Union (EU), Japan, and the United States is also provided (see Appendices A and B).
Pharmaceutical Quality and Manufacturing Control Issues
Cell Sources and Raw Materials
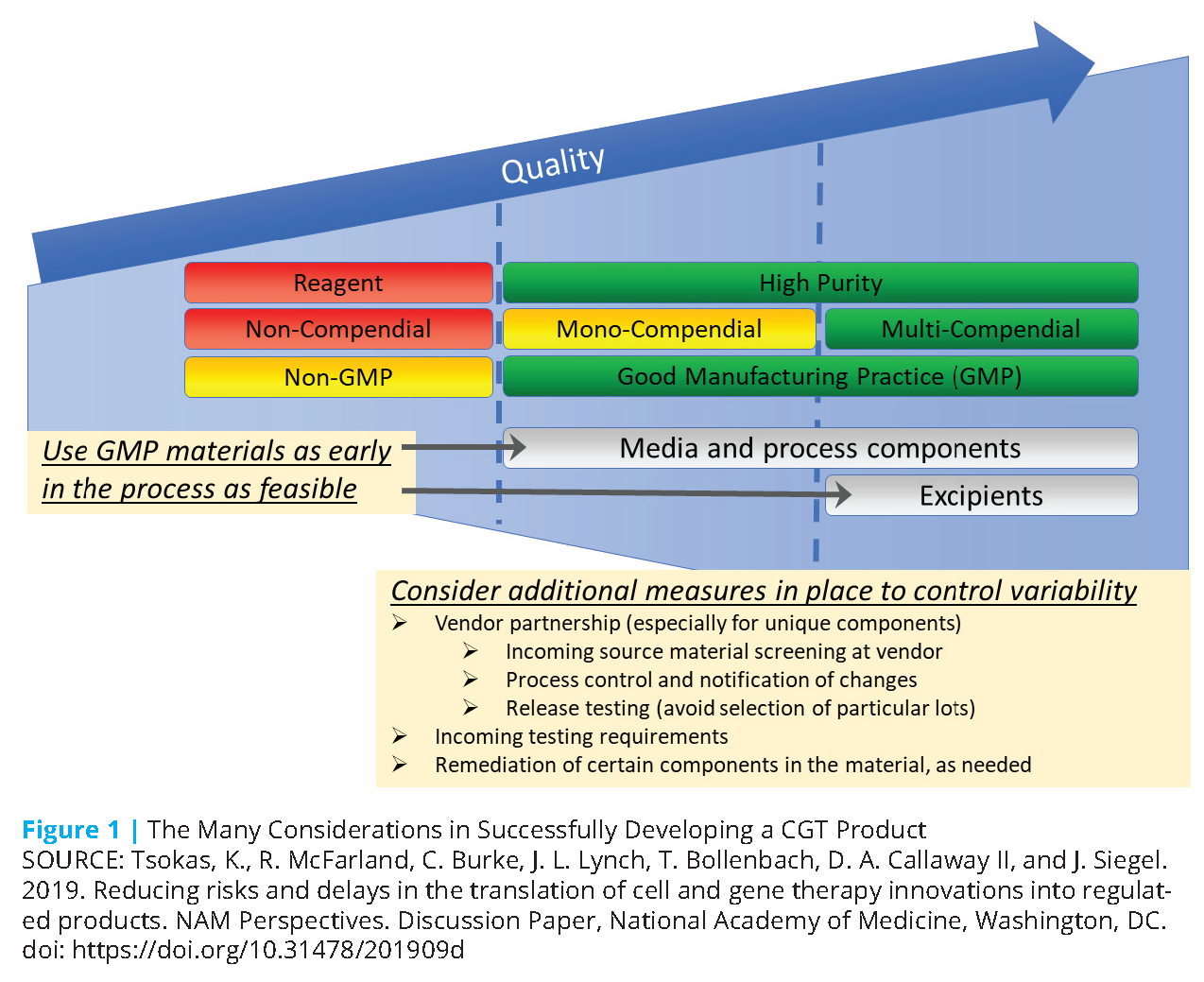
From the beginning of product discovery, developers can employ a careful and informed approach to the selection and use of preparation process materials to avoid future problems in development (see Figure 1). For cell therapies, such materials include both the source cells themselves and any other raw materials (referred to as “ancillary materials” in the EU, and may also be referred to as “components”) used in the process. Raw materials constitute one of the key elements of quality systems and are distinct from the drug substance, drug product, and—when applicable—device component. Progenitor cell banks not intended for further manufacture (e.g., working cell bank) may be classified as a drug substance. Cell banks used for other purposes (e.g., lentiviral vector production) may be classified as a raw material.
Cells
The cell sources for both autologous and allogeneic cell products are critical components of development planning. Patient-derived donor cells represent one of the most variable components in the manufacturing process for an autologous cell therapy product. Variability arises from intrinsic (e.g., genetic) and extrinsic (e.g., health status and medications) factors characteristic of the donor. For example, T cell distributions can vary normally from person to person. Quantity and distribution of T cell types in autologous starting material can be influenced by the patient’s health, disease progression, and the number and intensity of prior patient treatments.
All too often, critically important discovery science and early product development use primary cells or cell lines obtained from an unknown or untraceable donor. They also often use cells or cell lines from a donor who has been inadequately screened and tested for potentially transmissible agents as outlined in Food and Drug Administration (FDA) guidance (21 C.F.R. 1271, Subpart C—Donor Eligibility). Such uses lead to serious challenges down the road, including challenges in the ability to administer the product.
Employing a donor eligibility determination minimizes the risk to product handlers and recipients and reduces, or identifies, variables that might affect product quality. Per FDA guidance, cells intended for autologous use must be labeled “For Autologous Use Only” (21 C.F.R. 1271.90[c][1]) and “Not Evaluated for Infectious Substances” if donor screening is not performed (21 C.F.R. 1271.90[c][2]) [1]. Thus, screening for infectious agents may be suggested by FDA, even though it is not yet part of required FDA exclusion criteria. For products that use patient material as a starting material to create a master cell bank/working cell bank as the final product, donor testing should be aligned with 21 C.F.R. 50 [2], 56 [3], and 1271 [4] and EU Directives 2004/23/EC [5] and 2006/17/EC [6].
While donor characteristics cannot be fully standardized for autologous therapies, uniform procedures can be applied in subsequent treatment of the donor material, thereby reducing product variability. With more experience, it may be possible to identify markers or patient specifications that impact the manufacturing process or final product characteristics. However, hospitals and institutions may have different proprietary processes for collecting (e.g., aphaeresis machines), processing, and freezing autologous donor cells, leading to differences in procedure. Some differences may be a result of available equipment and established handling procedures already in place.
Monitoring of emerging standards from the Foundation for the Accreditation of Cellular Therapy (FACT) (http://www.factwebsite.org/Standards/) and other standards entities will be valuable as the field develops. For example, FACT accreditation will ensure that institutional practices follow quality standards for collecting and handling samples, which are expected to improve the consistency of source materials. In the early development and potential transfer of the process, equipment and practices should be standardized as much as possible. Personnel should be trained in standardized procedures for collecting and processing cells to provide additional control of source cell variability.
For allogeneic cells, a well-documented history and characterization of the cells are imperative, since they will be the source of a master cell bank. A comprehensive history documenting testing—including donor eligibility determination, source materials, and passage history—is highly recommended.
Raw Materials
The complex needs and high demand for many component materials in the development of CGT products present some challenges for raw material sourcing. Although use of many sources of raw materials may be acceptable in the preparation of early-phase trial material, selecting an appropriate vendor and material grade can avoid delays, potential failure, and additional costs in later development, especially if a new vendor and material would eventually need to be selected and qualified. Developers should use clinical and pharmaceutical-grade materials when available and, as is feasible, consider cost of goods sold (COGS). These are not regulatory requirements but are recommended.
Consistency and reliability of raw materials are critical to ensuring consistent manufacturability and comparability of product through development. Country-specific compendial and regulatory requirements—including those from United States Pharmacopeia (USP), European Pharmacopoeia, and Japanese Pharmacopoeia—and other relevant requirements should be understood and addressed early in development. Some materials may be in high demand and have limited suppliers, so vendor capability and stability should be considered when selecting component materials (e.g., media, beads, assay reagents, disposable sets). Also, the number of vendors that supply the raw material can be a factor in developing a sustainable process. Due to the complexity or proprietary nature of some reagents, only one supplier may be available (sole-sourced components). In other cases, more than one vendor can supply the material, but only one supplier is either acceptable or qualified (single sourcing).
Although sole and single sources are common in early development, every attempt should be made to select materials that can be sourced from multiple vendors to enable more flexibility and ensure a reliable supply in later development and the commercial stage. The critical components will typically require more attention to sourcing and characteristics than other more standard components of the process. For allogeneic cell source (apheresis) materials, both chain of custody and chain of identity must be established and maintained throughout the entire production process.
Academic discoverers and developers should bear in mind that when the manufacturing process is transferred out of a clinical research or hospital setting, the receiving facilities may not have the same medical equipment or ready access to some materials. Thus, the process may need to be changed. Developers should avoid equipment or materials that may not be readily available and engage in advance planning for transfer (e.g., validating and procuring substitutes) to prevent major development risks or delays.
Because some components may be novel or not commonly used, both the supplier and developer may have limited experience in the manufacture of these components and in the definition of their key quality attributes. Research-grade proteins may contain tags or impurities not desirable for human administration, and human-derived components require extensive screening to ensure viral safety. Custom disposable sets, reagents, and media are examples of complex components that may require particular attention to the reliability of their manufacturing, quality, and consistency.
The proprietary nature of some components can also lead to complications and delays in development. If the supplier of a raw material is unwilling to share full information about the manufacturing, composition, and attributes of the material with the CGT developer, the developer may still use that material. In FDA filings, the developer would cross-reference a manufacturer’s master file, which will contain confidential information filed with the FDA but not disclosed to the developer. However, such an approach has the potential to hamper CGT product development and impair understanding of any impact of raw material variability on the CGT product. Lack of such information prevents the developer from providing input regarding control of the raw material process and/or attributes. Also, the lack of material understanding will impact the ability to assess removal throughout the process, will affect the ability to evaluate the associated risks of residuals and impurities, and may constrain process investigations.
Selection of clinical, USP, or development grades of materials also can impact future development. Although lesser grades of materials may be acceptable for use in early development, process development leading to definitive clinical trials and ultimately commercialization will require higher grades (e.g., USP). Although this system is not a grade, it is desirable that raw materials are prepared in accordance with Good Manufacturing Practices (GMP), if such materials are available. Doing so ensures control over the manufacturing environment, contributing to greater lot-to-lot consistency. If reference materials exist, they should be used. The availability and stability of compendial grades of raw materials should also be considered when selecting these for development. Developers need to understand the impact of critical quality attributes (CQA) and quality requirements for each raw material.
As more materials are GMP sourced and development proceeds to later phases, the COGS will increase due to the additional testing and controls required for GMP-grade materials. Increased volumes in later development or initial launch may not necessarily provide cost relief if the raw materials are in high demand or there are limited vendors. Although COGS may not be considered important for early development, choices of raw materials at this stage may have a significant influence on the COGS of the final product. Selection of proprietary components or sole-sourced materials may restrict cost minimization, and issues with production or availability of these materials could lead to clinical and commercial delays. Selection of proprietary equipment may also lock the developers into using sole-sourced components, such as associated disposable sets and reagents (such as affinity beads), which can contribute to a high COGS. Proprietary components and processes may also carry additional royalty costs, which will further increase the cost of the final drug product.
For genetically modified cell products, the transduction and transfection processes can add significantly to the overall COGS. The use of viral vectors, such as lentivirus, significantly adds to the cost, especially if high quantities are needed to achieve the desired level of transduction. In these cases, attention should be given to maximizing transduction efficiency to avoid significant expense from the use of large quantities of the vector.
Although the cost of viral vectors may be avoided by using nonviral transfection methods or gene editing techniques, reagents and intellectual property considerations of those approaches may contribute significantly to the overall cost of the cell product. Selection of an option early in the process, with these considerations in mind, can result in fewer changes and faster progress in later development.
Attempts should be made to ensure the input materials and process do not introduce particles into the final product. In addition to implementing a closed process, developers should select materials and ancillary supplies with low particle burden to minimize requalification and potential delays later in development.
Since subvisible particles cannot be reliably detected and quantified in the presence of cells, an aseptic process simulation can be performed with all the media and components in the process but without the cells themselves. The aseptic process simulation may help to identify potential issues with specific components, material vendors, or operations. To mitigate risk for introduction of particles into the final product, where possible, developers should select vendors that exercise particle control procedures and conduct appropriate testing for their materials. Thorough documentation of particle observations early in development will aid in the particle strategy as the program proceeds.
Product Quality and Consistency
For complex biological systems, it is challenging to establish and maintain consistency of the product throughout development stages and in the final commercial process. Multiple factors affect their viability and function. Failure to ensure consistency of the product can lead to costly problems when promising early results are not readily reproduced. The ability to produce a consistent product will depend on controlling critical process parameters (CPPs) and monitoring CQAs, along with other factors that define the overall quality of the product. Consistency will also be a part of establishing comparability between processes to allow for optimization of the process.
Early versus Late-Phase Requirements: Qualification and Validation of Analytical Methods
Development of analytical methods for CGT products, as with other biotherapeutics, involves increasing requirements as the candidate product progresses through clinical development phases. While the regulatory requirements for discovery and early development may be limited, detailed product characterization with reliable methods early on can yield great benefits not only in terms of development of qualified assays, but also in terms of, even more importantly, determination of the product CQAs (those test results that correlate with desired activity and quality). For example, early incorporation of in-process testing facilitates incorporation of quality-by-design principles into process development. This incorporation will then facilitate the scaling up of manufacturing and the troubleshooting of out-of-specification products.
For early phase analytical methods, assay qualifications will be phase-dependent and may not meet all International Conference on Harmonization (ICH) Q2R1 Validation of Analytical Procedures criteria [7]. However, these methods should at least be qualified for the intended use, and the equipment should have gone through the appropriate qualifications (IQ/OQ/PQ) to limit equipment variability in the resulting analytical data.
Although available analytical methods may be limited at the outset, early phase specifications must be designed to ensure safety of the product. Characterization assays are common with early phase release and stability, as a way to gather data and determine the final analytical testing strategy of the product. Collection of robust characterization data early in development can help better define critical attributes and demonstrate comparability for changes later in development. Employing in-process monitoring and testing early on will be valuable for process understanding as development proceeds.
Potency assays in early phase development may be limited, as the mechanism of action (MoA) may not be well established and clinical materials may also be limited. As early as is feasible, a clinically relevant potency assay, surrogate, or matrix approach should be established to link potency to the product’s MoA. Early development of multiple assays of potentially relevant product activities will not only facilitate development of a potency assay, but will also help ensure that the product is consistent and that early results are relevant for designing later studies and for licensure. Further, a suitable potency assay provides valuable information concerning overall product stability and is useful for establishing comparability post-manufacturing process changes. In the United States, there are potency-specific guidance documents (see Appendix A). Other regions may require individualized scientific advice to fully establish the potency testing strategy for the product.
As development proceeds, analytical methodology and specifications should be evaluated and refined based on development data, engineering runs, and clinical batch analysis. Additional assays may also be added to monitor and control critical attributes of the product. ICH Q2R1 should be followed for late-phase assay validations.
As the clinical program reaches pivotal trials, analytical methods and specifications should be based on a sufficient dataset to tighten the release and stability specifications in support of late-phase comparability and process-validation activities. Health authority expectations increase considerably at this stage, and Pharmaceutical quality and manufacturing control becomes a critical focal point of product development. This component is analogous with the term “chemistry, manufacturing, and controls,” used for small molecules.
Stability
Whereas most CGT products are more sensitive to ambient temperature than other biotherapeutics are, careful early attention to stability under various processing and storage conditions is critical to developing a process that will generate a consistent quality product for development. The concern for stability extends beyond the final cellular products to the intermediate products, including the master and working cell banks and apheresis products. In addition, many of the ancillary materials may also be biologics or other chemicals with activities that can be diminished by exposure to adverse environmental conditions.
Autologous Products
For autologous cell products, the durations of different temperature exposures should be tracked starting from the collection of material and should include processing procedures and freezing and thawing methods. Having consistent handling, storage, and freezing procedures at the collection center will be important to understanding the stability characteristics and ensuring predictable yields across individual, patient-specific lots.
Maintenance of cryogenic temperatures during shipping will likely be critical to ensuring stable source material. When storage needs to be -65°C or higher (even if for brief periods during transfers or handling), the stability of the material should be characterized under such conditions. Developers will need a thorough characterization of the temperature exposures to ensure product integrity (e.g., cumulative time at -65°C experienced by the cell product).
Conditions for thawing the source cells and final CGT product should also be characterized and controlled to provide similar conditions between lots.
Allogeneic Products
Similar considerations for stability hold for allogeneic CGT products. An allogeneic product will generally not have a separate collection procedure for each lot, but factors affecting storage and handling of master and working cell banks will impact the stability of the cells and the initial manufacturing process steps. Exposures during the processes for producing the cell banks, cell intermediates, and the cell product should be controlled and kept consistent between lots. The limited stability of many cells outside of cryogenic conditions will restrict process times and exposure temperatures. Cells with limited stability will often require procedural considerations to minimize these exposures.
With allogeneic CGT products, developers will also need to ensure stability of the cell banks and product over a long period, typical of biotherapeutics. In addition, creating larger batches of allogeneic products may result in longer times at ambient temperatures before batch cryogenic freezing is performed. As a result, these process and stability losses may be greater than when working with autologous products, which are typically cryopreserved more quickly on an individual basis.
Comparability
The leveraging of clinical data throughout development can occur only if the drug product has similar or comparable qualities throughout development. Thus, comparability for CGT therapy products constitutes a critical aspect of process development and is required to link clinical data throughout progressive phases of product development. ICH Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process provides a basis for comparability that relates to biotechnology products, which can also be leveraged for regenerative medicine product development [8]. In addition, the recent FDA draft guidance Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) provides the FDA’s thinking in terms of comparability [9].
Challenges of limited batch volumes and variability of materials and assays can necessitate strategies for demonstrating comparability that are different from those used with typical biotherapeutics. Giving attention to the many aspects of comparability early in development will ensure that knowledge from early lots can be leveraged as the product enters later development. If there is no comparability plan available near the outset, data generated in early development may need to be repeated for later development and clinical bridging.
As stated in the recent FDA draft guidance, “the extent of comparability testing will depend on the manufacturing change, the ability of analytical methods to detect changes in the product, and the stage of clinical development” [9]. Elements of changes requiring comparability assessment include changes in source cells, reagents, components, process, formulation, and place of manufacture. Knowledge of product CQAs and development of sensitive assays to measure them are extremely important in demonstrating comparability.
In establishing comparability, assays that can be related to the MoA (see also the section below, “Potency Assay”) will be especially efficacious, particularly if they are robust regarding reagent availability and variability. Reliance on orthogonal assays adds more confidence, especially if existing assays are highly variable or if the potency assay does not fully reflect a known MoA.
Depending on the complexity of the CGT asset and the importance of comparability to the future success of the CGT asset, companies should plan to engage with FDA at Initial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) meetings, pre-Investigational New Drug (IND) meetings, and formal FDA meetings as product development continues. Developers addressing comparability without health authority engagement proceed at risk (i.e., without prior FDA engagement), which can lead to delays when the developers do adapt to FDA feedback and/or repeat comparability.
Being consistent with source cells, raw materials, and process components will help in comparability determinations. Consistency of the process and lot-to-lot comparisons may be difficult to establish if materials in the process are changed and the amount of material is limited. If developers ensure a suitable amount of single lots of materials to cover many CGT product lots, they will better understand comparability and control other sources of potential variability.
Lastly, although with autologous programs the traditional drug substance and drug product definitions often do not fit well with the manufacturing process, the concepts that underlie the terms regarding the existence of retention samples need to be incorporated in the manufacturing process. Therefore, companies working on autologous programs should have a drug substance and drug product retain policy in place, supported by clinical documentation, to allow patient samples to be used for research. These retain samples are critical for method development and comparability exercises as development continues.
Potency Assay
As mentioned earlier, reliable measures of a product’s potency early in development can minimize delays and failures as the product moves through development. Potency is a quantitative or qualitative measure of the biological function of a cell therapy, gene therapy, or engineered tissue and is a reflection of the anticipated or observed clinical efficacy. Assays measuring product potency are of central importance to all product development and quality control activities. Potency assays serve many functions, including helping to identify and control sources of variation in product activity, and testing for product comparability across lots, process, site, scale changes, and stability testing as part of a comprehensive analytical control strategy. Given the complexity of CGT products, demonstrating consistent potency is critical. Therefore, development and use of potency assay(s) should begin very early in product development.
In anticipation of licensure and eventual product commercialization, potency assays must comply with current GMP regulations and therefore need to be quantitative, measure product-specific biological function, have predefined acceptance and rejection criteria, and be validated. They should be able to be performed rapidly, especially when being used for the release testing of products with a very short shelf life. Early in development, there may not be sufficient information to define criteria or to fully validate, but the potential to achieve those standards should be present. Although potency can, in practice, be evaluated in vitro or in vivo, a quantitative in vitro test, whether cell-based or not cell-based, is highly desirable. Potency assays should represent the product’s MoA and should correlate to a likelihood for clinical efficacy, even if the correlation is understood incompletely.
Establishing the link between the potency assay and MoA can be particularly challenging for product development teams because CGT products are multifactorial and sometimes have undefined MoAs. However, as Bravery and colleagues point out, the expected MoA of the cells or tissues comprising the final construct or formulation should at least be partially known if the product is being considered for clinical development [10]. To mitigate risk during early stages of product development, a matrix of assays targeting several hypothetical MoAs should be explored and then refined as product knowledge increases, particularly during clinical evaluation.
Hypothetical MoAs can be generated from literature searches—screening experiments, known properties, and activity of the cells—and from any previous clinical experience. In addition to testing hypothetical MoAs, discovery-based technologies, including multiple omics platforms, can be used to generate potential surrogate markers of potency. Even after one MoA is determined to be most critical and becomes the basis for a potency assay, assays of other activities may nonetheless remain useful in evaluating and ensuring product consistency.
Potency assays are typically implemented progressively. As stated above, a matrix of assays targeting several hypothetical MoAs and surrogate potency markers identified through discovery platforms should be explored early in development. Typically, these assays will have acceptance criteria with very broad ranges, reflecting a relatively low level of product understanding and limited manufacturing experience. The potency assays in the matrix, and their respective acceptance criteria, should be constantly evaluated as clinical studies and product understanding progress. Therefore, the correlation between clinical efficacy and potency should increase.
Developers should start potency assay development early by collecting as much data as possible to characterize the product and how it works. As with manufacturing process development, the need for extensive and focused product characterization during early phase development cannot be overemphasized. During development, it is critical that extensive but focused product development activities be balanced against the desire to progress quickly, as a hurried approach carries significant risks during scale-up and commercialization. In the long term, a hurried approach can result in manufacturing processes that have tight constraints and little flexibility, which may compromise commercial success. For example, without a method for measuring potency, even minor manufacturing process or material changes could require expensive clinical qualification [10].
General Evaluation of Uniqueness of Analytical Assays
Because of numerous factors, analytical assays are unique. These factors include the youth of the field regarding manufacturing at scale; the diversity of regenerative medicine indications; the heterogeneity of source materials; the ingenuity of potential products which contributes to a diversity of products and to a concern for retaining intellectual property; and the consequence of global regulatory frameworks predicated on the evaluation of the details of each specific product and clinical indication. There are currently many analytical assays used in the field. These assays are often developed by the same groups developing the therapeutic product for which the assay is used.
In general, there is much less risk to the program if protocols and reagents are widely used and readily available. Using common reagents and protocols will increase the likelihood of successful assay transfers by limiting additional variables, such as reagent source and handling differences. Expanding the range of some critical material attributes may increase flexibility in the sourcing or cost of goods. However, a thorough understanding of the impact on the product CQAs will be required. Specialized assays and those that require unique reagents or more state-of-the-art equipment may be desirable (or even required) to more fully elucidate cell characteristics. However, such reagents and equipment usually are accompanied by scarce assay history and potentially higher variability. In some cases, extremely novel assays may require use of a comparability plan to ensure effective bridging to more conventional assays.
Some industry groups—such as the Alliance for Regenerative Medicine and regulators such as the European Medicines Agency and the FDA—recognize that the proliferation of assays without standardization is costly. Recently, assay standardization efforts have gained momentum as a result of the increase in approvals for CGT therapies. These standardization efforts have also been bolstered by the FDA’s newly given authority—in section 3036 of the 21st Century Cures Act of 2016—to work with the National Institute of Standards and Technology to define standards for regenerative medicine therapies.
The Standards Coordinating Body—whose mission is to “[c]oordinate the accelerated advancement and improved awareness of the standards and best practices that address the rapidly evolving needs of the global regenerative medicine advanced therapy community”—won an FDA contract in 2017 to implement the provisions of section 3036 [11]. If successful, these efforts could significantly de-risk development across regenerative medicine by reducing the expense of developing all testing and analytical tools on an ad hoc basis and by improving the reliability and reproducibility of assays used.
Specificity
Specificity is critical in ensuring that sample components will not interfere in an assay readout. Especially in the case of autologous cell therapy, the variability of the input material, and sometimes the process, can affect the cell product and may lead to interference with some assays. However, if developers establish acceptance criteria at the time of collection, they can mitigate this variability. For autologous cell preparations, there may be significant cell variability lot-to-lot, so data will need to be accrued and analyzed to better understand the impact of different cell populations on assay specificity. Changes in the health or treatment history of the patient donor, process changes, or changes in raw materials may also impact specificity of the assay. The effect of these changes should be monitored throughout development to ensure the assay readout truly reflects the activity of the cells of interest and not activity of other cell populations or excipients.
Process Controls
If developers define and control the process early, they may avoid problems applying early lessons and reproducing early results as development proceeds. Because CGT products incorporate numerous raw materials, including human cells that are sensitive to myriad biochemical and physical stimuli, understanding what to control constitutes a major challenge to ensuring a stable process. Selection of appropriate reference standards can also be challenging. To address these challenges, the FDA has continued to refine its guidance on process control and validation. The FDA, most recently, adopted quality-by-design principles of continuous improvement under an initiative titled Pharmaceutical CGMPs for the 21st Century—A Risk-Based Approach [12] and in ICH Q8 for pharmaceutical development [13]. The quality-by-design approach, although not mandated by the FDA for CGT therapies, should be given strong consideration by developers of these types of products, as it is a promising approach to mitigating the significant risk posed by complex manufacturing processes.
Development of a well-controlled manufacturing process should begin with a set of predefined quality characteristics that are linked to the safety and efficacy of the product, and that define what the process intends to generate. Typically, these attributes are summarized in a living document called a Quality Target Product Profile.
Developers use the Quality Target Product Profile to generate a list of product CQAs. A CQA is defined as a “physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality” [13]. Examples of quality attributes for CGT products include purity, potency, identity, and sterility—and in the case of engineered tissues, examples include structural or mechanical properties. To determine the criticality of these quality attributes, developers should subject each one to a risk assessment and to correlative preclinical or clinical studies to assess its impact on product safety and efficacy.
The attributes of raw materials and manufacturing process parameters that may vary and can therefore impact CQAs are known as critical material attributes (CMAs) and CPPs, respectively. Important attributes with variability that might impact product quality, CMAs and CPPs should be measured and controlled to ensure that the process generates a product of the desired quality [13]. The degree to which each identified CMA and CPP impacts CQAs is often evaluated initially in a risk-based analysis and subsequently by Design of Experiments (DOE)-based studies. The DOE studies screen for potential variability and assess the impact of CMAs and CPPs on the quality of process intermediates (e.g., cells, scaffolds, vectors) or on the final product. Measurements that quantitate the impact of variability on CQAs can be developed using methods similar to those described for the development of potency assays. These methods include the collection of large amounts of characterization data from discovery-based multiple-omics platforms.
Nonclinical Issues
The nonclinical safety program is an important but often underemphasized aspect of product development. Developers conduct nonclinical testing to provide evidence of possible therapeutic benefit for patients, to provide information on potential toxicities, and to identify at what doses these effects manifest. The nonclinical safety program prior to FIH clinical trials is critically important to ensuring that the product progresses from therapeutic development and into clinical studies in a timely and cost-effective manner. Because CGT products are complex, researchers may not be aware of regulatory recommendations and requirements associated with preclinical testing. Several regulatory guidance documents (which can be found in Appendices A and B) can help guide the design of a nonclinical program. However, developers of each CGT product will need to conduct different studies to understand the safety liabilities of their own products. Cell- and gene-based therapies will often require a unique nonclinical program to justify testing in humans, and consequently, sponsors should seek early advice from a regulatory authority (e.g., INTERACT, pre-IND, scientific advice).
With regenerative medicine products, often the majority of the nonclinical program can be conducted prior to FIH testing. Due diligence review with partners and collaborators can happen at any time during a program. Throughout the field, the trend is to complete such a review toward completion and prior to the FIH package. It is important to have a sound strategy for the implementation of the nonclinical safety program to prove to both regulators and investors that the product is safe to administer to patients. This section will focus on the preclinical safety work that should be conducted prior to FIH clinical trials.
Developers should remember that the preclinical safety package is conducted in appropriate animal models to help them understand and de-risk the potential toxicities to human patients, and to acknowledge that no preclinical model is perfect. Each model will have strengths and weaknesses. It is important to understand these limitations and seek regulatory advice if any questions arise when forming the species selection strategy and translating the results for the FIH package.
The following sections will address some common pitfalls encountered when planning a nonclinical strategy.
Species Selection
Choosing a toxicology species constitutes one of the most critical aspects of the nonclinical safety program. Species selection forms the foundation of the preclinical toxicology program and associated regulatory strategy. If an appropriate species justification is not provided, additional studies will need to be conducted, delaying the clinical program and coming at a significant cost. It is a common misperception that biologics should be tested in a nonhuman primate model or that two species are needed for the development of CGT products.
Most importantly, the species should demonstrate a biological response similar to that in humans. For gene therapies, this means that the activity of the protein transcribed by the transgene binds the target with affinity that is reasonably close to the human and elicits a similar pharmacologic response. Additionally, for in vivo administration, the animal model needs to be permissive for transduction with the viral serotype being used [14]. For cell therapies, the presence of the pharmacologic target is critical, but the influence of the microenvironment on cellular products and the hurdle of the immune response to xenotransplantation need to be considered. Often only one species is pharmacologically relevant, but that single species is sufficient to support an FIH program. No regulatory requirement exists for two species for CGT products.
When choosing the toxicology species, developers should also consider the route and method of administration. For many CGT products, local delivery to the target area may be required; however, it may not be feasible to test the safety of the route of administration in smaller species (mice and rats). In these cases, if a rodent model is used for toxicity studies, a second, larger species may be required to study the safety of the route of administration.
Due to the high degree of species specificity of these products, along with the xenotransplantation immune response (graft vs. host disease), developers often use nontraditional animal models to characterize the safety of the products. Benefits and limitations of nontraditional animal models are discussed below.
Genetically Engineered Animals
The use of genetically engineered animals (knock-out, knock-in, and transgenic) often constitutes a useful approach for CGT products. Most commonly, developers use immunocompromised mice (athymic and nude, and severe combined immune-deficient strains) to overcome the immune response from xenotransplantation. Humanized knock-in animals or transgenic animals are also used when the pharmacologic target is only expressed in humans or the disease or injury state. Use of genetically modified animals facilitates risk assessment when a standard toxicology model is not relevant; however, limitations accompany these models. In general, there is a lack of historical data for these models, which then necessitates a full characterization of the model [15]. Such characterizations may involve running a mock study prior to conducting a study with the CGT product to understand findings associated with the model, route of administration, and age of the animals. Additionally, it may be prudent to add control animals to the study. However, obtaining substantial numbers of genetically engineered animals may be challenging.
Animal Models of Disease
An animal model of disease may be the most appropriate species for preclinical development due to the features of CGT products. For example, the target may be expressed or not expressed only in a disease state, or some safety concerns may arise only when there is an interaction between the CGT product and a disease environment. Additionally, these studies can improve clinical dose selection and therapeutic index by providing insight into the relationships of dose to activity and toxicity [16]. As with genetically engineered models, there is often a lack of historical data on animal models of disease states. These animals also often have a limited life span and may be difficult to produce and obtain, leading to limited numbers of animals accessible for conducting the study.
Additional challenges with these models include the confounding effects from the disease and ability to identify what findings are caused by the disease state and what findings are caused by the therapeutic. Also, these disease models can be very complex, and multi-component processes (e.g., inflammation) may differ substantially between the disease model and humans. All of these factors lead to the need to fully characterize the model and understand the limitations from inherent variability as outlined by the FDA, including limited historical data, technical limitation with physiologic and anatomic constraints, animal care issues, and limited reliability in modeling human pathophysiology [17]. Without a good understanding of the variability and background lesions, it can be difficult to interpret test-article-related lesions, potentially leading to additional studies to understand specific findings.
Surrogate Products
An animal-derived product analogous to the advanced therapeutic product (surrogate) may be the most appropriate testing strategy. Using such a product will allow for collection of safety data in a species in which no pharmacologic activity exists in a more standard toxicology model (e.g., rodent, dog, cynomolgus monkey, mini-pig) that has a robust historical control and lacks the limitations in life span and sourcing. However, this product differs from what will be administered to humans. Consequently, characterization of both products (human therapeutic and surrogate) will be required to understand product impurities and, more importantly, comparability in function. Data will need to be obtained on the potential differences between the surrogate and the intended human product, including differences in bioactivity, molecular mechanisms, and role of the microenvironment [18]. Additionally, it is not appropriate to use the surrogate for tumorigenicity studies. These studies should be conducted only with the intended human product.
In Vitro Approaches
There are instances in which there are no in vivo models available to adequately assess safety liabilities. For example, there are T cell receptor T cell products that are human-specific due to major histocompatibility complex restriction. Also, as outlined above, there are limitations to approaches using animal models of disease and surrogates. It is important to use all the information and tools available when assessing the safety of a product. Doing a thorough target liability review, and including any previous clinical experience with similar products, any published experiences with the same target, and any nonclinical and clinical experiences with the cell types, vectors, and transgenes can provide valuable information that can be addressed early in product development.
Using in vitro methodologies to understand the expression profile of the target (e.g., in silico analysis, reverse transcriptase polymerase chain reaction, immunohistochemistry) and testing the specificity of the product via using various cell lines, primary cells, or iPSC-derived 3-D cultures from various tissue sources can help build confidence in the safety and potential liabilities of a product. (Induced pluripotent stem cells are called “iPSCs.”) Additionally, organoid models or organs on a chip may provide safety information. However, it is important to ensure that the microenvironment in these systems is appropriate to support the CGT product. While in vitro approaches can provide valuable information to support a CGT, developers should understand that they likely will not answer all safety questions. Such safety issues include biodistribution (cell migration to nontarget areas or tissues), risks of the delivery procedure, and potential inflammatory or immune response to the administered product.
If developers understand the liabilities associated with the CGT product, both in vivo and in vitro tools can be used to develop an adequate safety strategy to support FIH clinical trials.
Designing a Preclinical Program to Support FIH
Conducting a preclinical assessment of the safety of the advanced therapeutic product can help frame the acceptable risk-benefit ratio for the FIH clinical trial. This assessment needs to identify, characterize, and quantify potential toxicities and explore the reversibility and the effect of dose level for these findings [17]. The preclinical strategy for every product is unique. Developers should consider the proposed clinical indication and treatment plan, published preclinical and clinical safety data, and the pharmacology and intrinsic properties of the product. The following sections outline some points to contemplate when designing a program to support an FIH study.
Good Laboratory Practices
Standard pharmaceutical toxicology programs run through a series of non-Good Laboratory Practices (GLP) and GLP studies with increasing length to test a maximum tolerated dose. Developers characterize toxicity in more robust GLP studies. According to 21 C.F.R. 58, developers should conduct all preclinical studies in compliance with GLP. However, some components may not be available at a GLP testing facility, including the use of alternative animal models (genetically modified animals, animal models of disease)—along with capabilities to formulate and administer CGT products or capabilities to evaluate specific end points, such as vector distribution or cell fate. In these cases, it is appropriate to conduct toxicology studies or toxicology end points on efficacy studies in a non-GLP manner. However, these studies and end points need to use a prospectively designed protocol with reporting of deviations and amendments, and the data need to be collected and reported with sufficient quality and integrity to support the FIH program. Developers should ensure careful documentation of the study and preservation of raw data to avoid needing to repeat the study should it be required for a regulatory submission.
General in Vivo Study Design Considerations
In designing the in vivo toxicology study, developers should pay careful attention to selection of the test article, dose, route, schedule, and duration to support the proposed clinical trial design.
The test article is generally the CGT product, but as noted above, it may be a species-specific surrogate with similar potency and activity in the test species. The test article should be well characterized and test results should be documented. Excipients, particularly if they are noncompendial, may have their own toxicities that need to be defined.
The dose route, levels, and schedule should reflect the intended clinical dosing regimen. The method of administration (e.g., delivery devices used) should be the same as, or appreciably similar to, those proposed for the clinic. Developers should consider the safety of the route and method of administration (e.g., rapid intravenous administration of cellular products may lead to pulmonary complications; use of some needles or catheters may lead to shearing and loss of viability, resulting in irregular dosing).
Multiple dose groups should be included in the study to understand the relationship between dose and associated toxicities. The dose groups should include adequate numbers of animals per gender, randomized to each group and appropriate control groups (e.g., formulation vehicle control, sham surgery). Dose levels of the therapeutic need to be included to bracket the proposed clinical dose range, including the anticipated efficacious dose and exposure multiples of this dose or the maximum feasible dose.
The study needs to be designed to understand acute and transient effects and chronic effects. Standard safety parameters should be monitored and comprehensive pathology performed. Additional tests should reflect potential product or target-specific concerns, e.g., specific immunotoxicology end points, engraftment, vector distribution, and CGT product fate.
Multiple dose studies are needed only if the developers intend for the product to be readministered in the clinic. Developers should also include multiple sacrifice time points, along with the potential for recovery, to define acute and chronic toxicities. The biodistribution study can help inform the selection of study duration and sacrifice time intervals.
Tumorigenicity
The FDA examines the tumorigenicity evaluation prior to human testing of a CGT product. It may take up to two years to plan, pilot, and conduct the study, and generate a final report. Thus, tumorigenicity must be considered early in the discovery and development process to avoid clinical trial delays.
A tumorigenicity study tests the potential of a cell population—either in suspension or incorporated into a tissue construct—to display tumorigenic, hyperplastic, or dysplastic behavior at the site of administration and/or, through cell migration, at a distant site. In its guidance on deciding on the type and design of study, the FDA recommends a risk-based, product-specific approach. The potential risk of tumor formation should be determined through comprehensive product characterization and an understanding of the raw materials used in manufacturing the product, including the type, origin, and total number of cells contained within the product. The following factors should also be considered:
- The total number of cells administered (dose);
- Duration of culture and number of cumulative population doublings, as cells cultured to high population doublings may accumulate mutations that can lead to tumor formation;
- The origin and type of cells used to generate the product, as pluripotent stem cells may form benign teratomas upon injection, while terminally differentiated or specialized primary cell types should not;
- The differentiation stage of cells in the final product and the efficiency of conversion from pluripotency to progenitor or terminally differentiated cell type, or from one cell type to another;
- Whether the product is intended and likely to persist transiently and induce a temporary effect, or if the cells or tissue are intended to engraft and produce a durable effect;
- The distribution and migration potential of the cells; and
- Whether the cells are genetically modified.
Meta-analysis of clinical trials administering mesenchymal stem cells through various routes suggests that these cells also present a low risk of tumorigenicity [19]. On the other hand, because of their potential to form teratomas or tumors due to undifferentiated cells that remain in the final product, pluripotent stem cells (e.g., embryonic stem cells or iPSCs) and cell types derived from pluripotent stem cells are still viewed as having a concerning risk of tumorigenicity after implantation.
Potential tumorigenicity should be considered, depending on the level of perceived risk, the product’s attributes, the scientific literature, previous clinical experience, and in vitro assessments. In vitro studies can include characterization of biological stability, including proliferation rate and number of population doublings before senescence, and karyotyping or other genetic analysis to look for chromosomal abnormalities.
In the highest-risk cases, it may be necessary to carry out in vivo tumorigenicity assessments. In designing a study, careful consideration should be paid to the animal model, the monitoring period, the route of administration, and method of monitoring. In some cases, these assessments can be performed as part of toxicity studies, by examining individual animals for nodules. Regardless of the type of study, developers should generate the cells to be tested through a process that is as close as possible to the process being used to generate clinical samples.
In the positive control group, there should be strong tumorigenic activity in the species being tested. In the case of cells derived from embryonic stem cells or iPSCs, the donor stem cells themselves can act as positive controls, as both are capable of benign tumor formation. Developers can also include serial dilutions of the positive control in the final product to provide an indication of the threshold level of undifferentiated cells within the final product that will result in the formation of a tumor or other ectopic tissue.
For the assay to be most sensitive to tumor formation, both the animal model and the monitoring period need to be considered. Because human cells and tissues are being evaluated, the animal model should be immunodeficient to allow survival of the implant for a sufficient period. Typically, developers use immunodeficient rodents (e.g., nude, Severe Combined Immune Deficiency (SCID)). Preferably, developers should choose an animal type without high spontaneous tumor rates. Depending on the planned route of administration in humans, rodent models (in which subcutaneous testing is most commonly done) may be suboptimal. SCID pigs might be considered, as it is desirable to deliver the test article to the clinical site of delivery using a method similar to the one to be used in the human trial. The in vivo monitoring period should be at least nine to 12 months long, or for the typical life span of the species being used.
Pilot studies are often recommended to test the variables described above in combination. In particular, the dose of positive control cells—administered via the clinical route of administration, for a particular monitoring period—should be optimized for the target animals. Kawamata and colleagues summarize the considerations for the design of tumorigenicity studies for iPSC-derived cell therapies [20].
The initial sign of tumor formation can either be visual or perceived through physical examination in the case of gross tumor development, ectopic tissue formation, and other formations. Secondary analyses can include histology to determine the gross morphology of the tumor and IHC to determine the proliferative state of the tumor. Analyses should also include either genotyping or polymerase chain reaction of human-specific genes to determine whether the tumor originated from the implanted cells or tissue. A commonly used marker for detection of human DNA is the Alu gene, which can easily be amplified by polymerase chain reaction and shows a high sensitivity.
Intellectual Property
Many scientists and physicians engaged in the discovery and early development of CGT products have only cursory knowledge and awareness regarding IP. Some may value publication and dissemination of knowledge above the potential for financial gain. However, the development of a CGT product—both the manufacturing process and clinical trials—can be very costly. If careful attention has not been paid to protecting IP, there may be no incentive for investment in these costly aspects of development, and even the most promising therapies may never reach patients.
In general, IP can be thought of as any product of the human intellect protected by law from unauthorized use by others. Limited monopolies in protected property are inherently created due to intellectual property ownership. Most intellectual property falls into one of four categories: patent, copyright, trademark, and trade secrets [21]. These four types of IP are subject to different legal recourses and precedent under federal or state law, the details of which are beyond the scope of this article. However, a fundamental need exists for developers to consider protection of all IP prior to publication and/or pubic presentation of data. Once IP is disclosed, it cannot be undisclosed, and then it may be too late to protect it.
In addition to IP protection, careful attention to IP ownership can be critical to ensure that a viable development path exists and to protect the value of invested time and resources. Universities and established companies have policies that determine IP ownership. When founding a start-up, ownership of IP among the founders should be clearly agreed upon. Significant IP may be known and/or generated by consultants early in the life of a spin out, and it is important that ownership of that IP be fully and firmly understood. Use of open-source software may be practical for early spinouts but could result in downstream IP problems if the programs are inappropriately incorporated into a manufacturing process.
IP issues constitute one of the more common concerns for investors, and having a well-designed IP strategy can be a pivotal factor in the success or failure of a start-up company stemming from academia. These plans should be tailored by a professional, because the details of each specific situation and the optimal balance of disclosure, IP sharing, and IP protection is different for every firm. That balance should be well understood and defined a priori, as it may be expensive or impossible to address the issues later.
Communication and Technology Transfer
To avoid delays and inefficiencies during the development of a CGT product, developers should have clear development and regulatory paths and be able to communicate them clearly to colleagues, health authorities, and potential partners. A target product profile provides the overall strategic vision for the particular CGT product. Developers can use the profile to focus strategic questions during development and with health authority interactions to ensure clarity and alignment regarding regulatory requirements. As developers consider partnering with other companies or transferring CGT products to later-stage development and marketing, the profile, early regulatory strategy, and consistent and documented health authority interactions will increase the chances of efficient movement of the target product to production and release.
Communication of technical information (technology transfer) is critical whenever partnering and collaboration occurs, and particularly when responsibility for a project moves from an academic or corporate environment to another company. Technology transfer should be planned in advance, well understood, and carefully executed. As with IP, tension exists regarding the advantages of sharing information, the leveraging of partners’ or vendors’ experience, and the retention of confidential information. Even if the communication or transfer does not contain protected IP, how much and when technology is transferred can drastically affect the perceived and real value of the company to potential investors or corporate suitors. Communication of technical information should comply with the rules of financial regulators, such as the US Securities and Exchange Commission. For an investigational product regulated by the FDA, the type and timing of the disclosure can be critical.
Summary
CGT products have the potential to address many unmet medical needs. They are often discovered and initially developed in academic laboratories or small biotechnology companies with expertise in science and innovation. Due to their lack of expertise in drug development, these groups may inadvertently choose approaches that lead to avoidable cost increases, delays, or other problems later in development.
This paper reviews key issues that may arise during development of CGT products and suggests potential strategies to minimize risks and allow for easier translation from research into later stage development and marketing. The paper focuses on pharmaceutical quality and manufacturing control, nonclinical testing, intellectual property, communication, and technology transfer issues—all relevant from a global perspective. However, developers should also address regional requirements when developing CGT products.



Join the conversation!
![]() Tweet this! Some medical innovations, like cell and gene therapy products, face a difficult path to regulation. A new #NAMPerspectives discussion paper outlines some possible risks and how to avoid them: https://doi.org/10.31478/201909d
Tweet this! Some medical innovations, like cell and gene therapy products, face a difficult path to regulation. A new #NAMPerspectives discussion paper outlines some possible risks and how to avoid them: https://doi.org/10.31478/201909d
![]() Tweet this! This new #NAMPerspectives discussion paper outlines a pathway for easier translation of cell and gene therapy research into product development. Read more: https://doi.org/10.31478/201909d
Tweet this! This new #NAMPerspectives discussion paper outlines a pathway for easier translation of cell and gene therapy research into product development. Read more: https://doi.org/10.31478/201909d
![]() Tweet this! Innovations like cell and gene therapy that are discovered in smaller labs and companies often have trouble developing to market stage. Read more in this #NAMPerspectives paper on how risks and delays can be avoided in this process: https://doi.org/10.31478/201909d
Tweet this! Innovations like cell and gene therapy that are discovered in smaller labs and companies often have trouble developing to market stage. Read more in this #NAMPerspectives paper on how risks and delays can be avoided in this process: https://doi.org/10.31478/201909d
Download the graphic below and share it on social media!

References
- 21 U.S.C. § 1271.90 (2019). Available at: https://www.ecfr.gov/cgi-bin/text-idx?SID=02c3b3874f51842f020b5987a92b59eb&mc=true&node=se21.8.1271_190&rgn=div8 (accessed February 15, 2019).
- 21 C.F.R. § 50 (2019). Available at: https://www.ecfr.gov/cgi-bin/text-idx?SID=02c3b3874f51842f020b5987a92b59eb&mc=true&tpl=/ecfrbrowse/Title21/21cfr50_main_02.tpl (accessed February 15, 2019).
- 21 C.F.R. § 56 (2019). Available at: https://www.ecfr.gov/cgi-bin/text-idx?SID=02c3b3874f51842f020b5987a92b59eb&mc=true&tpl=/ecfrbrowse/Title21/21cfr56_main_02.tpl (accessed February 15, 2019).
- 21 C.F.R. § 1271 (2019). Available at: https://www.ecfr.gov/cgi-bin/text-idx?SID=02c3b3874f51842f020b5987a92b59eb&mc=true&node=pt21.8.1271&rgn=div5 (accessed February 15, 2019).
- European directive 2004/23/EC (2004). Available at: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:102:0048:0058:en:PDF (accessed February 15, 2019).
- European directive 2006/17/EC (2006). Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006L0017&from=EN (accessed February 15, 2019).
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). 2005. Validation of analytical procedures: Text and methodology, Q2(R1). Geneva, Switzerland. Available at: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed February 15, 2019).
- ICH. 2004. Comparability of biotechnological/biological products subject to changes in their manufacturing process, Q5E. Geneva, Switzerland. Available at: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf (accessed February 15, 2019).
- U.S. Food and Drug Administration (FDA). 2018. Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drug applications: Draft guidance for industry. Washington, DC. Available at: https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM610795.pdf (accessed February 15, 2019).
- Bravery, C. A., J. Carmen, T. Fong, W. Oprea, K. H. Hoogendoorn, J. Woda, S. R. Burger, J. A. Rowley, M. L. Bonyhadi, and W. Van’t Hof. 2013. Potency assay development for cellular therapy products: An ISCT review of the requirements and experiences in the industry. Cytotherapy 15(1):9-19. https://doi.org/10.1016/j.jcyt.2012.10.008
- Standards Coordinating Body. 2019. Overview. Available at: https://www.standardscoordinatingbody.org/overview-mission (accessed October 17, 2018).
- FDA. 2004. Pharmaceutical CGMPs for the 21st century—a risk-based approach. Washington, DC. Available at: https://www.fda.gov/media/77391/download (accessed February 15, 2019).
- ICH. 2009. ICH Harmonized Tripartite Guideline: Pharmaceutical Development, Q8(R2). Geneva, Switzerland. Available at: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf (accessed February 15, 2019).
- MacLachlan, T. 2013. Turning the corner with viral-based gene therapy—development of the rogue biopharmaceutical. In Nonclinical development of novel biologics, biosimilars, vaccines and specialty biologics, edited by L. M. Plitnich, and D. J. Herzyk. Amsterdam, Netherlands: Elsevier. Pp. 259-285.
- Todd, M. D., and M. Dempster. 2013. Regulatory guidelines and their application in the nonclinical evaluation of biological medicines. In Nonclinical development of novel biologics, biosimilars, vaccines and specialty biologics, edited by L. M. Plitnich, and D. J. Herzyk. Amsterdam, Netherlands: Elsevier. Pp. 35-64.
- Bussiere, J. L., P. Martin, M. Horner, J. Couch, M. Flaherty, L. Andrews, J. Beyer, and C. Horvath. 2009. Alternative strategies for toxicity testing of species-specific biopharmaceuticals. International Journal of Toxicology 28(3): 230-253. https://doi.org/10.1177/1091581809337262
- FDA. 2013. Guidance document: Preclinical assessment of investigational cellular and gene therapy products. Washington, DC. Available at: https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM376521.pdf (accessed February 15, 2019).
- Bailey, A. M., M. Mendicino, and P. Au. 2014. An FDA perspective on preclinical development of cell-based regenerative medicine products. Nature Biotechnology 32(8):721-723. https://doi.org/10.1038/nbt.2971
- Lalu, M. M., L. McIntyre, C. Pugliese, D. Fergusson, B. W. Winston, J. C. Marshall, J. Granton, and D. J. Stewart. 2012. Safety of cell therapy with mesenchymal stromal cells (safecell): A systematic review and meta-analysis of clinical trials. Public Library of Science One 7(10):e47559. https://doi.org/10.1371/journal.pone.0047559
- Kawamata, S., H. Kanemura, N. Sakai, M. Takahashi, and M. J. Go. 2015. Design of a tumorigenicity test for induced pluripotent stem cell (IPSC)-derived cell products. Journal of Clinical Medicine 4(1):159-171. https://doi.org/10.3390/jcm4010159
- Cornell Legal Information Institute. No date. Intellectual property. Available at: https://www.law.cornell.edu/wex/intellectual_property (accessed February 15, 2019).